Here we differentiate between BOS and other phenotypically overlapping syndromes.
- Interstitial chromosome 20q11.21 micro deletion
- Novo overlapping of chromosome 20q11.2 duplication
- Partial deletion of ASXL1 gene
- Novo truncation ASXL3 mutation or Bainbridge-Ropers Syndrome
- Shashi-Pena Syndrome – De Novo truncation Variants in ASXL2
- Opitz C Syndrome
- KLHL7 or Perching Syndrome
Interstitial chromosome 20q11.21 micro deletion
The (novo nonsense) mutation in the ASXL1 gene occurs in Bohring-Opitz Syndrome (Hoischen et al. 2011). So a part of the gene is changed. On the other hand, an interstitial chromosome 20q 11.21 micro deletion is a chromosome mutation in which a part of the chromosome is missing.[1] This is in contrast to a change (mutation) of a single gene (part of a chromosome). The micro deletion on chromosome 20q 11.21 contains the gene ASXL1 gene.
Only six cases with an interstitial 20q11.21 microdeletion have been reported. Patients with this micro deletion have some symptoms which are similar to BOS: mild intellectual disability (and speech delay), facial dysmorphisms (microretrognathia, upslanting palpebral fissures, prominent eyes, broad nasal bridge, low set ears, and low frontal hairline, hypertelorism, cleft palate), feeding problems (feeding intolerance and food refusal), failure to thrive, heart defect, preaxial polydactyly (right thumb) and retinal dysplasia (Iourov et al. 2013, Posmyk et al. 2014 and Jedraszak et al. 2015). All the facial dysmorphisms and feeding problems are observed in cases of 20q11.2-q.12 loss but also in novo ASXL1 mutation. Patients with novo ASXL1 mutation usually exhibit more severe symptoms (i.e. severe/profound intellectual disability; growth retardation; and craniofacial, ophthalmic, and neurological abnormalities) than patients with an interstitial microdeletion on 20q11.21 chromosome (Iourov et al. 2013). Interestingly all the presented patients do not exhibit trigonocephaly and the typical BOS “posture”.
The conclusion of the study by Iourov et al. (2013) is that a change or loss of the ASXL1 gene, causes typical BOS appearance and characteristics. A change (mutation) is much more serious than a loss (microdeletion) of a particular gene, in this case the ASXL1 gene. They add that the presence of an ASXL1 mutation in their case is unlikely.
Until 2014 the interstitial microdeletion of the 20q11.21 chromosome is not connected to a specific disease. Whether the microdeletion of the 20q11.21 chromosome should be associated with BOS or whether it should be its own disease has not been decided then. While Iourov et al. (2013) suggested that the interstitial micro deletion of the 20q11.21 chromosome should become a contribution to the phenotype BOS as an example of a nonmutated ASXL1 loss, Posmyk et al. (2014) proposed to form another group because the children are very similar to each other and are distinguishable from other BOS children in terms of disease progression.
The existence of a 20q11.2 microdeletion syndrome has been proposed, based on five previously reported cases. Today the study of Jedraszak and colleagues (2015) support that 20q11.2 microdeletion syndrome is a new contiguous gene deletion syndrome with a recognizable phenotype. (Jedraszak et al. 2015)
Bohring et al. (2012) concluded during the European Human Genetics Conference 2012 in Germany: “Further carefully performed genotype-phenotype studies are necessary to specify the most appropriate key symptoms and to differentiate between BOS and other phenotypically overlapping syndromes or to prove genetic heterogeneity.”
So far, it has not been published that a hereditary case has ever been identified. The FB group has been informed that there is a case of this disease being passed to 2 siblings from their maternal grandmother via their mother.
Novo overlapping of chromosome 20q11.2 duplication
Copy number gains of ASXL1 occur in chromosome 20q11.2 duplication syndrome and cervical cancer. Truncation mutations of ASXLs occur in autism, Bohring-Opitz and related syndromes (Katoh 2015). Avila et al. 2013 suggest a novel microduplication syndrome for four patients with overlapping chromosome 20q11.2 microduplication. They show that such patients display common clinical features including metopic ridging/trigonocephaly, developmental delay, epicanthal folds and short hands. The duplication comprised the ASXL1 gene. Because of craniofacial features in common with Bohring-Opitz syndrome, in particular metopic ridging and trigonocephaly, they suggest also that duplication of ASXL1 contributes to the phenotype.
Partial deletion of ASXL1 gene
In our Bohring-Opitz Syndrome support group on Facebook we know a child with partial deletion of ASXL1 gene. The SNP (single nucleotide polymorphism)[2] chromosomal microarray testing has shown that the deletion on chromosome 20 is 165 kb in size and is located in 20q11.21. It contains the last 9 exons of the ASXL1 gene.
We have learned that a novo mutation of the ASXL1 gene causes Bohring-Opitz syndrome (Hoischen et al. 2011), and that an overlapping chromosome 20q11.2 microduplication comprised the ASXL1 gene and it contributes to the phenotype (Avila et al. 2013). Furthermore, the interstitial 20q11.21 microdeletion (contains the ASXL1 gene) lead to some symptoms which are similar to BOS (Iourov et al. 2013 and Posmyk et al. 2014).
In this particular case it is uncertain if the clinical significance of a partial deletion of ASXL1 caused BOS because the child didn’t fit the main characteristics of BOS. The geneticist suggest that this case is more comparable with a (larger) microdeletion of the 20q11.21, which is described by Iourov et al. (2013) . They propose that the loss of the gene is the main cause of the patients differences. They also mention that it is possible that a deletion or partial deletion causes a much milder form of Bohring-Opitz Syndrome.
The child, born in breech position, is diagnosed with a macrocephaly with an external hydrocephalus that has not dissipated. Apart from “differences in facial features” with skin colouring difference between eyes and on back of head the child was furthermore diagnosed with mild dilation of her aortic heart root. In addition the child has a mild to moderate hypotonia and hip dysplasia. Oral and verbal apraxia as well as severe oral aversions and sensory issues have affected eating, talking and some fine motor since birth. Although the child is very smart it has a hard time showing this due to its limited verbal abilities. Chronic slow transit constipation and severe bloating issues along with slower digestion were noticed. However, since the age of 5 these issues seem to get better. The child is small for its age, but not to an extend which may cause concerns at this point. The child is delayed in all milestones and for this reason it takes part in physical and occupational therapy as well as feeding and speech therapy. The child crawled at 12 months and walked at 20 months. The child was described as very smart and sweet natured with a very laid back happy personality.
Novo truncation ASXL3 mutation or Bainbridge-Ropers syndrome
The ASXL3 gene is mapped to the 18q12.1 chromosome. ASXL3 belongs to the same gene family as ASXL1 gene and ASXL2 gene. Mutations in ASXL3 appear to be associated with a disorder that is similar to BOS (Dinwiddie et al. 2013). All of these mutations occurred de novo in the penultimate exon, in a region analogous to that in ASXL1 where mutations resulting in Bohring-Opitz syndrome occur (Bainbridge et al. 2013).
There are 15 children presented with the de novo nonsense and frameshift mutation in the ASXL3 gene in four studies (Bainbridge et al. 2013 and Dinwiddie et al. 2013, Srivastava et al. 2016, Hori et al.2016 and Kuechler et al. 2016). The children have phenotypes similar to BOS, including severe feeding difficulties, failure-to-thrive and neurologic abnormalities with significant developmental delay. Further, they showed less phenotypic overlap with patients who had de novo truncating mutations in ASXL1 (Bainbridge et al. 2013). The conclusion from Bainbridge et al. (2013) is: “We have identified truncating mutations in ASXL3 as the likely cause of a novel (new) syndrome with phenotypic overlap with Bohring-Opitz syndrome.” The name is Bainbridge-Ropers syndrome (BRS). [3]
While no patient from the study of Bainbridge et al. (2013) had the typical ‘BOS posture’ of elbow and wrist flexion, or myopia or trigonocephaly, the patient presented by Dinwiddie et al. (2013) does have trigonocephaly. Thus, the lack of trigonocephaly may not be useful in differentiating BOS from this newly recognized condition. To date, none of the fifteen patients described with ASXL3 mutations has displayed the typical BOS posture, whereas 100 percent of the 30 described patients with ASXL1 mutations did, suggesting that this feature might be useful in discriminating between the two related conditions (Dinwiddie et al. 2013).
In the latest article on Bainbridge-Ropers Syndrome Kuechler et al. emphase the need of further delineation of the clinical phenotype of BRS, but by presenting six newly diagnosed patient they consider “Bainbridge-Ropers Syndrome as distinct entity […] which is distinguishable from Bohring-Opitz Syndrome”. The patients with BRS do show characteristic craniofacial phenotype so as long face with prominent forehead, arched eyebrows with mild unibrow, downslanting palpebral fissures, prominent nose bridge, small alae nasi, high, narrow palate and relatively little facial expression with open mouth appearance (Kuechler et al. 2016).
Parents of children with Bainbridge-Ropers Syndrome have set up the Facebook group Bainbridge-Ropers Syndrome. This group, dedicated to families with children who have Bainbridge Ropers-Syndrome and ASXL3 genetic mutation, is a place to share stories, give advice, post updates of new developments, add photos, and most importantly to offer support to each other. More information on BRS can be found on their website.
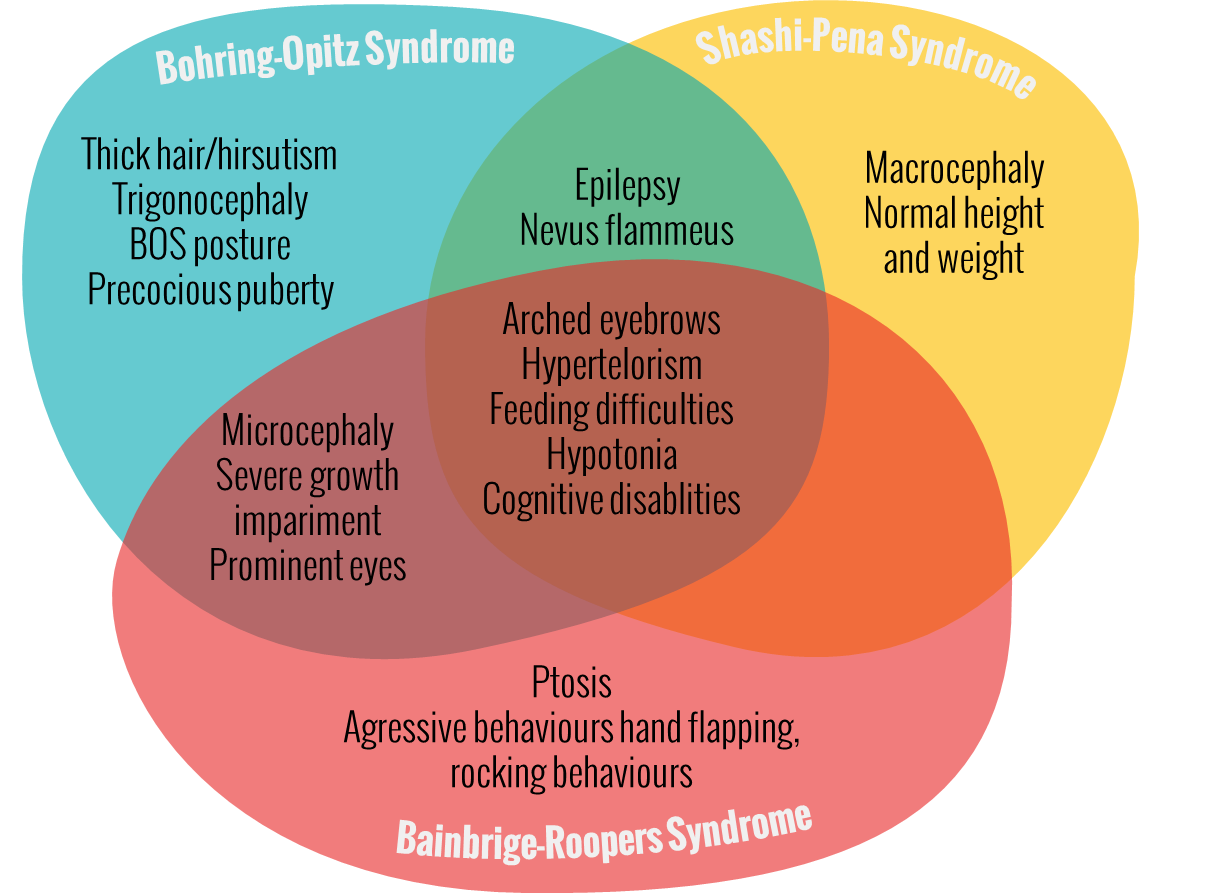
Shashi-Pena Syndrome – De Novo Truncation Variants in ASXL2
Vandana Shashi, a professor of pediatrics for the Division of Medical Genetics at Duke University School of Medicine, and colleagues present the first patient with De Novo Truncating Variants in ASXL2. Five additional children, all with the same physical features and the ASXL2 gene mutation were found throughout GeneMatcher. However the new disease has no name yet, Shashi said: “We can now definitively say this is a newly identified disease, [… .] With just one case, we could not say the gene mutation was the underlying cause. But with six cases, all with the same ASXL2 mutation, it is definitive.”[4] ASXL2 belongs to the same gene family as ASXL1 gene and ASXL3 gene. Mutations in ASXL1 and ASXL3 have been respectively implicated in causing Bohring-Opitz Syndrome (BOS) and Bainbridge-Ropers syndrome (BRS) with overlapping symptoms of severe intellectual disability and dysmorphic features but more severe impairments than patients with De Novo Truncating Variant in ASXL2. These six patients have overlapping characteristics with Bohring-Opitz Syndrome, like prominent eyes, arched eyebrows, hypertelorism, a glabellar nevus flammeus, neonatal feeding difficulties and hypotonia. Different from BOS and BRS patients with ASXL2 (Shashi-Pena Syndrome) mutation have features like macrocephaly, absence of growth retardation and more variability in the degree of intellectual disabilities.[5]
Currently the Undiagnosed Disease Network (UDN) is looking for patients with variants in ASXL2 gene.
Opitz C syndrome
Opitz C Syndrome (OCS) shared many features with the Bohring-Opitz syndrome (BOS). Several experts have proposed the possibility that, in fact, they could be a single syndrome with a variable spectrum of severity from the mildest Opitz C Syndrome too the more severe BOS.
The Opitz C Syndrome or C Syndrome, also known as Opitz Trigonocephaly Syndrome, is a rare and very severe disease. It is estimated to affect one child in a million. But the high mortality (50% of patients die within the first year of life) of this syndrome and the difficulty to correctly diagnose it led various experts to suggest that this may be a more common syndrome, more frequent among undiagnosed miscarried foetuses and stillborn babies.
The most characteristic features of this malformation syndrome characterized by trigonocephaly are severe mental retardation, hypotonia (poor muscle tone), variable cardiac defects, redundant skin, and dysmorphic facial features such as squat, flat nose, very narrow eyes and at an inclined angle, epicanthal folds, low and rotated ears, including upslanted palpebral fissures (summary by Kaname et al., 2007)[6]. Joint contractures are also very common, limiting the patients’ mobility, and they often also have a certain degree of internal organs affectation (cardiac and renal malformations…). It is also common among these patients to suffer from hypotonia (poor muscle tone) and seizures.
The gene responsible for this disease are unknown. Indeed, it appears that there are more than one gene involved. To date, there have been found mutations associated with these syndromes in only 2 genes. On one hand, half of the patients with BOS bear mutations in the ASXL1 gene, being clearly involved in the disease. Instead, only two patients (out of 27 analysed) bear changes in CD96 gene. More recently it have been observed that patients without any resemblance to the OCS or BOS syndromes but with renal diseases have truncating mutations at the CD96 gene, which raises serious doubts about the role of this gene in BOS and OCS.
Identification of the gene or genes responsible for a disease is the first step to understand it, which, in turn, represents the starting point in the development of therapies.[7]
NORD National Organisation for Rare Disorders offers information on C Syndrome.
KLHL7 or Perching syndrome
Perching syndrome (KLHL7) shares many features with the Bohring-Opitz syndrome (BOS). Perching syndrome is an autosomal recessive multisystem disorder (OMIM #617055) caused by homozygous or compound heterozy-gous variants (truncating and missense) in Kelch Like Family Member 7 gene (KLHL7), located on chromosome 7p15. The acronym PERCHING has been proposed to include the most characteristic features and each letter represents two important phenotypic elements:
- P for Postural and Palatal abnormalities,
- E for Exophthalmos and Enteral-tube dependency/feeding issues,
- R for Respiratory distress and Retinitis pigmentosa,
- C for Contractures and Camptodactyly,
- H for Hypertelorism and Hirsutism,
- I for IUGR/growth failure and Intellectual disability/developmental delay,
- N for Nevus flammeus and Neurological malformations, and
- G for facial Gestalt/Grimacing and Genitourinary abnormalities (Jeffries et al., 2018).
This disorder is also referred to as KLHL7-related Bohring-Opitz-like phenotype due to overlapping phenotype. Because patients with a clinical diagnosis of BOS do not have mutations in ASXL genes further testing reveald the causing gene, and the review of published patients concluded that this disease could be clinically identifiable and perhaps considered a disease of its own. Although the phenotypic spectrum of Perching syndrome is broad and multisystemic, there seems to be a dysmorphic presentation that may allow the clinical recognition of this syndrome. Since the first report of this syndrome in 2016 (Angius et al., 2016), 18 patients have been reported.[10]
[1] (http://en.wikipedia.org/wiki/Deletion_(genetics): Types of deletion include the following: ‘Terminal Deletion’ — a deletion that occurs towards the end of a chromosome. Intercalary Deletion / Interstitial Deletion — a deletion that occurs from the interior of a chromosome. Microdeletion — a relatively small amount of deletion (up to 5Mb that could include a dozen genes). Microdeletion is usually found in children with physical abnormalities. A large amount of deletion would result in immediate abortion (miscarriage). 22. March 2015
[2] http://en.wikipedia.org/wiki/Single-nucleotide_polymorphism: A Single Nucleotide Polymorphism, also known as Simple Nucleotide Polymorphism, (SNP, pronounced snip; plural snips) is a DNA sequence variation. 22. March 2015
[3] When Dr Ropers from the Max-Planck Institute for Molecular Genetics in Berlin found a child with non-specific symptoms he used the genome-wide sequencing, researchers found that the child had a ‘truncating’ mutation in the gene ASXL3 not present in either parent. Bainbridge et al. (2013) first presented 4 unrelated patients with heterozygous nonsense and frameshift mutations in ASXL3 gene. 22. March 2015
[4] https://www.sciencedaily.com/releases/2016/09/160929133616.htm, 29.09.2016
[5] Shashi, VandanaBacino, A. et al.: De Novo Truncating Variants in ASXL2 Are Associated with a Unique and Recognizable Clinical Phenotype. The American Journal of Human Genetics DOI: http://dx.doi.org/10.1016/j.ajhg.2016.08.017
[6] OMIM 211750 April 4th 2015
[7] Biotech Spain: A race against the clock for the reseach at C Opitz Syndrome. News 23.12.2014. https://biotechspain.com/en/news_item.cfm?iid=opitz-c-crowfunding-ub, April 4th 2015
[8] Russell, B. E., Kianmahd, R. R., Munster, C., Yu, A., Ahad, L., & Tan, W.-H. (2023). Clinical findings in 39 individuals with Bohring–Opitz syndrome from a global patient-driven registry with implications for tumor surveillance and recurrence risk. American Journal of Medical Genetics Part A, 191A: 1050–1058. https://doi.org/10.1002/ajmg.a.63125
[9] Bruel A-L, Bigoni S, Kennedy J, et al. Expanding the clinical spectrum of recessive truncating mutations of KLHL7 to a Bohring-Opitz-like phenotype. Journal of Medical Genetics, 54(12), 830–835. https://doi.org/10.1136/jmedgenet-2017-104748
[10] Makay, P., et al. (2022). PERCHING syndrome: Clinical presentation in the first African patient confirmed by clinical whole genome sequencing. American Journal of Medical Genetics Part A, 1–7.
https://doi.org/10.1002/ajmg.a.62855
